Protein misfolding in neurodegenerative diseases
The function of most cellular proteins is dependent on their three-dimensional structure, which is acquired through folding of the amino acid chain. Understanding how proteins self-assemble into their specific structures rapidly and reliably is not only a fundamental problem in biology, but also of considerable relevance to medicine. Many proteins can “misfold” to create non-native structures. If these escape the cellular quality-control mechanisms, they can cause a wide range of diseases. In the case of several neurodegenerative diseases, the misfolded structures are able to propagate by converting properly folded molecules into more copies of the misfolded structure. Such behaviour was first identified in diseases like kuru and Creutzfeldt-Jakob disease in humans, scrapie in sheep, “mad-cow” disease, and chronic wasting disease in deer and elk, which are all caused by misfolding of the prion protein (PrP). More recently, however, “prion-like” propagated misfolding has been seen in other, more common neurodegenerative diseases, including Parkinson’s, Alzheimer’s, various other dementias, and ALS. All of these diseases are progressive, fatal, and lack effective treatment.
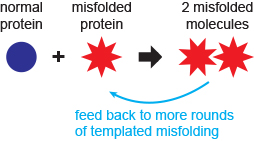
We are interested in understanding how proteins misfold and what determines the structural outcome. By directly observing single proteins as they fold and misfold, we attempt to answer such questions as: Can we directly observe misfolding events and misfolded states? How are they connected to the native folding pathway? What conditions increase the misfolding, or reduce it? In many cases, misfolded proteins aggregate into oligomers and even larger structures like amyloid fibres. What nucleates the formation of an oligomer? How does the oligomerisation proceed? How do different environmental conditions like temperature, metal ions, and pH change the aggregation? Can we find compounds that inhibit misfolding or aggregation, and how do they so? We investigate these general questions by looking at specific proteins related to disease. Currently, we are investigating PrP (prion diseases), α-synuclein (Parkinson’s), SOD1 (ALS), aβ (Alzheimer’s), and tau (Alzheimer’s and other dementias), using a combination of single-molecule assays (based on both fluorescence and force), ensemble biochemical approaches, and computational simulations.
Prion protein (PrP)

PrP is particularly notable as the first protein shown to form a misfolded structure that is “infectious,” in the sense that it can propagate misfolding and transmit disease. We aim to understand how mutations in PrP can change the propensity for prion diseases, whether increasing the likelihood in the case of mutations causing familial disease or decreasing the likelihood in the case of protective mutations, by examining how mutations change folding and misfolding at the single-molecule level. We also aim to probe the propagation of misfolding, by observing properly folded PrP molecules as they come into contact with misfolded molecules. A third goal is to probe the interactions of PrP with compounds that are known to inhibit misfolding or propagation, studying how they change the folding dynamics of PrP to determine their mechanism of action. Lastly, we aim to understand how “strains” of prions (slightly different versions of misfolded PrP) can propagate selectively and adapt to drug treatments by changing the mixture of strains present in the cell.
Superoxide dismutase 1 (SOD1)
SOD1 is an essential anti-oxidant enzyme whose misfolding is a prominent feature of ALS. We aim to understand how single SOD1 molecules misfold (the pathways followed and how they differ from native folding), the mechanism by which misfolded SOD1 can convert natively folded SOD1 to propagate misfolding, how pathogenic mutations change the misfolding and lead to different patient survival times, the effects of mutations that confer protection against disease, and the mechanism of action of compounds and antibodies that suppress misfolding.
α-Synuclein, Aβ, tau
These proteins differ from PrP and SOD1 by being generally disordered. Instead of having a well-defined native structure, they sample a variety of structures depending on conditions, often transiently. We aim to characterise the structures these proteins explore on their own and as they aggregate into small oligomers, which are thought to be the most neurotoxic species, using this knowledge to discover and test new compounds that can inhibit aggregation. We also aim to understand how these proteins interact with cellular chaperones that help to suppress aggregation under normal conditions.